In equation (7.1) it is simplest to treat the nuclei as fixed and so, the kinetic energy terms of the nuclei are not considered. In eq (7.2) the kinetic energy terms for the nuclei 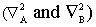 are not shown. This is known as the Born_Oppenheimer approximation and helps greatly in simplifying the problem. Equations (7.1) and (7.2) do not have any exact solution. We need to develop strategies to solve these as accurately as possible. In the present chapter we will develop qualitative ideas of the nature of the solutions. A few public domain (free) softwares are available for quantitative solutions. The Schrodinger equation for are not shown. This is known as the Born_Oppenheimer approximation and helps greatly in simplifying the problem. Equations (7.1) and (7.2) do not have any exact solution. We need to develop strategies to solve these as accurately as possible. In the present chapter we will develop qualitative ideas of the nature of the solutions. A few public domain (free) softwares are available for quantitative solutions. The Schrodinger equation for 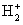 (a one electron system) has an exact solution, but that is in the bipolar coordinate system. We will instead consider numerical solutions here. (a one electron system) has an exact solution, but that is in the bipolar coordinate system. We will instead consider numerical solutions here.
|