There are two main observations in this figure 7.10(b). One is that the energies of the MOs are distance dependent and the other is that as the distance (r) between the nuclei is increased, the difference in the energies of the bonding and antibonding MOs diminish and vanishes as 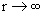 . At very large r, there is no binding and the energies are simply the energy levels of two separated non-interacting atoms. In fig 7.10(a) you will notice that the energy of the antibonding MO, . At very large r, there is no binding and the energies are simply the energy levels of two separated non-interacting atoms. In fig 7.10(a) you will notice that the energy of the antibonding MO, 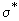 is greater than zero (the energy of the AO is taken as the reference value of zero) throughout. The reference value of zero is actually the energy of the atomic orbital (AO). The energy of the bonding orbital is lower than the energy of the AO for all but the very short distances. At very short distances, the energies of the MOs rise very steeply; far more steeply than 1/r, which is the formula for Coulomb repulsion between the electrons. The origin of this steep repulsion is not Coulombic, but the Pauli exclusion principle. When electrons are forced to be very close to one another, there arises a possibility that all the four quantum numbers of two electrons may be the same. Since this is forbidden by the exclusion principle, and furthermore since the electrons are indistinguishable as well (ie labeling the electrons as 1234 is no different from labeling them as 1432 since one can not associate any labels with these tiny indistinguishable particles), the energy rises sharply at short distances. is greater than zero (the energy of the AO is taken as the reference value of zero) throughout. The reference value of zero is actually the energy of the atomic orbital (AO). The energy of the bonding orbital is lower than the energy of the AO for all but the very short distances. At very short distances, the energies of the MOs rise very steeply; far more steeply than 1/r, which is the formula for Coulomb repulsion between the electrons. The origin of this steep repulsion is not Coulombic, but the Pauli exclusion principle. When electrons are forced to be very close to one another, there arises a possibility that all the four quantum numbers of two electrons may be the same. Since this is forbidden by the exclusion principle, and furthermore since the electrons are indistinguishable as well (ie labeling the electrons as 1234 is no different from labeling them as 1432 since one can not associate any labels with these tiny indistinguishable particles), the energy rises sharply at short distances. |