| |
On either sides of the equilibrium bond length r0, the PE rises as a symmetric quadratic function (a parabola). The vibrational wavefunctions can be obtained by solving the Schrodinger equation. The Hamiltonian operator (for energy) now consists of a kinetic energy term and a potential energy term V as shown in Fig. 13.4 and the solutions for energy, Ev have already been given in Eq.(13.20). The selection rules for the harmonic oscillator are:
Δv = ± 1 (13.22)
We will see several equally spaced lines (spacing hν) corresponding to the transitions 0→1, 1→2, 2→3 and so on. The first transition will be the most intense as the state with v = 0 is the most populated.
In actual diatomics, the potential is anharmonic. A good description of an anharmonic oscillator is given by the Morse function.
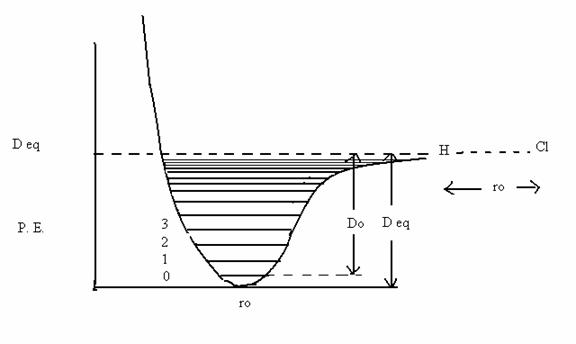
P.E. = Deq [1 – exp {a(ro-r }]2 (13.23)
In Eq. (13.22), Deq is the depth of the PE curve and r0 is the bond length. A plot of the Morse curve and the energy levels for the Morse potential are given in Fig. 13.5. The formula for the energy levels of this anharmonic oscillator is
Ev/hc = ev = (v+ ½) ν - (v+ ½)2 ν xe, cm-1 (13.24)
Here xe, is called the anharmonicity constant whose value is near 0.01. It can be easily deduced from the above formula that the vibrational energy levels for large υ start bunching together.
|
| |
Fig. 13. 5. The Morse potential and the energy levels therein. Note the difference between the dissociation energy Do and the depth Deq. All molecules have a minimum of the zero point energy of hν/2 corresponding to the ν = 0 state. This is a consequence of the uncertainty principle! |