3.10.3.2. Transition-State Inhibitors as Enzyme-Targeted Drugs
Transition State Stabilization by Enzymes: A catalyst bind the altered substrate in the transition state (T.S.) more tightly than it binds the substrate in the ground state (G.S.) whereupon lowering of the energy barrier of a reaction takes place. In the moment, lasting perhaps 1 msec, during which the catalytic event occurs, binding is enhanced by a factor that equals or surpasses the factor by which the catalyst enhances the rate of the reaction.
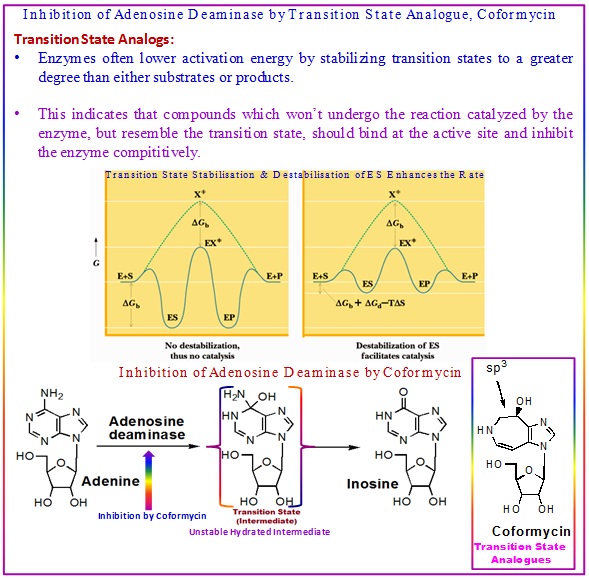
Figure 3.10: Mechanism of inhibition of adenosine deaminase by T.S. analogue Coformycin.
The transition-state analogues function as tight binding enzyme inhibitors. Till 1980s, most of the known examples of transition state analogues were natural products. Later on, in the 1990s, several synthetic inhibitors became the predominate examples of transition-state inhibitors. As for example, hydrolytic deamination of adenosine to inosine, catalyzed by fungal and mammalian enzymes (deaminase), is strongly inhibited by analogues of an Unstable Hydrated Intermediate formed by 1,6-addition of substrate water approaching from the front side of the adenosine ring. Thus, 1,6-hydroxymethyl-1,6-dihydropurine ribonucleoside (HDHPR) and the antibiotics coformycin and 2'-deoxycoformycin are powerful competitive inhibitors whose structures are very similar in structure to the postulated intermediate in the catalytic process.
Captopril, the first rationally designed enzyme-targeted drug, can be considered a transition-state inhibitor of peptidyl-dipeptidase A, also known as ACE. For HIV retropepsin (HIV protease), a variety of structure-assisted drug design techniques were successful in developing inhibitors. Saquinavir, the first marketed HIV retropepsin inhibitor, involved hydroxyethylamine isosteres that function as transition-state analogues.
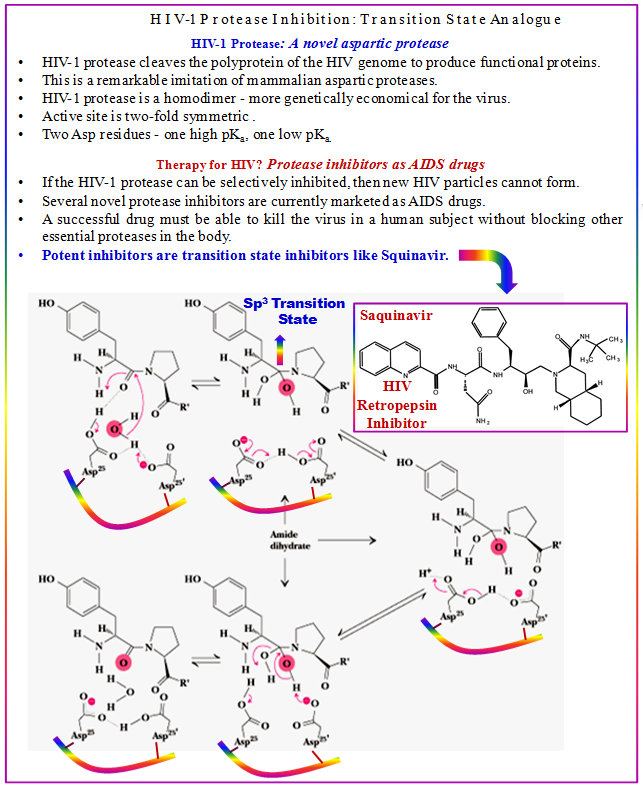
Figure 3.11: HIV-protease-I inhibition-a T.S. analogue approach.