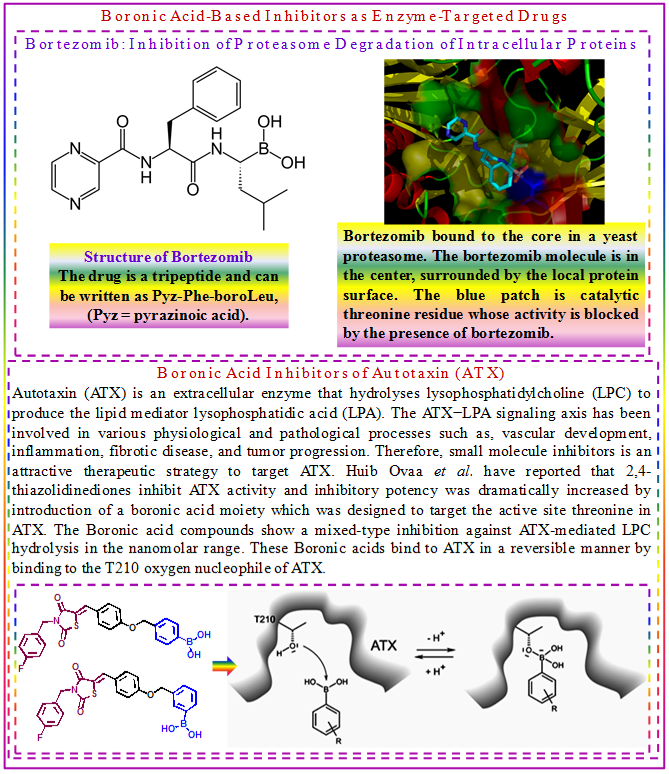
3.10.3.5 Boronic Acid-Based Inhibitors as Enzyme-Targeted Drugs
Boronic acids act as strong Lewis acids. The ready interconversion of a trigonal planar sp2 geometry to a tetrahedral sp3 geometry by substitution at boron is remarkably similar to the formation of tetrahedral intermediates in many hydrolytic enzymes, and allows boronic acids to act as reaction intermediate analogues with potent inhibitory properties. The first FDA-approved boronic acid-based inhibitor which came into the market in 2003 is Bortezomib which inhibits proteasome degradation of intracellular proteins and has shown efficacy in solid and hematological cancers. This is a slow-binding inhibitor.
3.10.3.6. Noncompetitive Inhibitors as Enzyme-Targeted Drugs
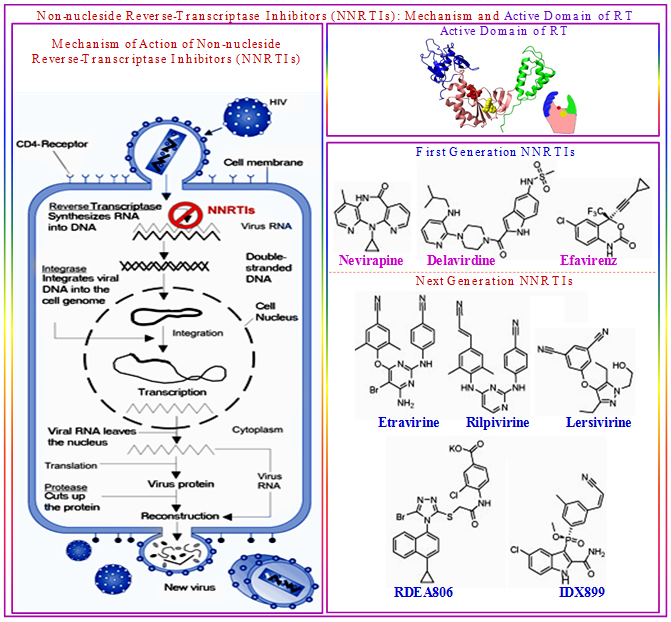
As we have seen the most enzyme-based drugs are active site inhibitors. However, noncompetitive inhibitors that do not bind in the active site may also show drug activity. Nonnucleoside reverse transcriptase inhibitors, including nevirapine, are a good example. Nonnucleoside inhibitors do not interfere with nucleotide binding or the change in conformation induced by nucleotide binding, but, they bind at an allosteric site of the enzyme, and thereby slowed down the rate of chemical catalysis. Therefore, the microscopic rate constants can provides a competitive advantage to enzymologists developing noncompetitive inhibitors as drugs. Two inhibitors might have the same Ki, but if one slows the rate of catalysis 10-fold more than the other, it will be a more potent drug.
3.10.3.7. Activators as Enzyme-Targeted Drugs
We know that drugs may activate or inactivate the enzyme activity. Till now we were discussing about inhibitors of pathogen’s enzyme as the drug to give a beneficial clinical effect to a patient. However, there are no pure enzyme activators on the marketaccording to a pure kcat or kcat/Km activator. However, recently, the concept of drug design target to develop enzyme activators (endogeneous enzyme) which might lead to a beneficial clinical effect in response to a certain disease and in future this approach may lead to a numbers of such kinds of drugs.
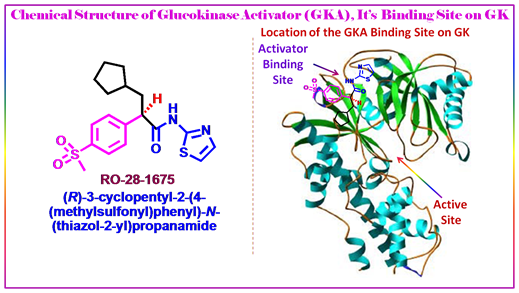
Figure 3.16: RO-28-1675 as glucokinase activator.
Joseph Grimsby et al., of Roche have recently discovered activators of glucokinase that increase kcat and decrease the S0.5 for glucose, and these may offer a treatment for type II diabetes. Glucokinase (GK) plays a key role in whole-body glucose homeostasis by catalyzing the phosphorylation of glucose in cells that express this enzyme, such as pancreatic β cells and hepatocytes. By screening of a library of 120,000 structurally diverse synthetic compounds, they found one small molecule that increased the enzymatic activity of GK. Chemical optimization of this initial molecule led to the synthesis of RO-28-0450 as a lead GK activator which is a class of antidiabetic agents that act as nonessential, mixed-type GK activators (GKAs) that increase the glucose affinity and maximum velocity (Vmax) of GK. RO-28-0450 is a racemic compound. Activation of GK was exquisitely sensitive to the chirality of the molecule: The R enantiomer, RO-28-1675, was found to be a potent GKA, whereas the S enantiomer, RO-28-1674, was inactive. RO-28-1675 also reversed the inhibitory action of the human glucokinase regulatory protein (GKRP). The activators binding in a glucokinase regulatory site originally was discovered in patients with persistent hyperinsulinemic hypoglycemi. The result of RO-28-1675 as a potent small molecule GKA may shed light to the chemical biologists to devise strategy for developing activators. Thus for a success to this end we must focus on highly regulated enzymes, or cooperative enzymes such as glucokinase, where nature has provided binding sites that are designed to modulate catalysis.