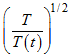A Simple Molecular Dynamics Program
Let us consider a simple MD program [Frenkel and Smit,2002] as given below

Figure 8 Pseudo Algorithm for Molecular Dynamics Calculation (Adapted from Frenkel and Smit, Understanding Molecular Simulations: From Algorithms to Applications. Academic Press.2002)
Initialization step
In this step the initial positions and velocities are assigned to all the particles in the system. The particles coordinates are chosen in such a way so that the molecular or atomic core overlap is avoided. The number of particles are also chosen keeping in mind the system of interest and its number density at a particular temperature. Usually a starting point is to choose a simple cubic lattice .Values of density and initial velocity are chosen so that the system becomes mechanically stable. Each particle is first assigned a lattice site and the velocity component of each particle is drawn from a uniform distribution such as Maxwellian in the interval [-0.5,0.5].The velocities are now shifted such that the total momentum is zero[Frenkel and Smit,2002]. The rescaling of the velocities are done so as to adjust the mean Kinetic energy to the desired value. It should be noted that the following relation holds:

Here![]() is the velocity in the α direction and m the mass of the particle.The instantaneous temperature at time t can be defined as:
is the velocity in the α direction and m the mass of the particle.The instantaneous temperature at time t can be defined as:

Thus comparing the equations (a) and (b), the instantaneous temperature T(t) can be matched with the desired temperature T by scaling the velocities by a factor of