1.10 Geometry Optimization
.....Quantum chemical computation starts with an optimized structure of the molecule. Structural changes within a molecule produce differences in its energy and other properties. The way the energy of a molecular system varies with small changes in its structure is specified by its potential energy surface. Geometry optimization locates the minima on the potential energy surface, thereby predicting equilibrium structures. At both minima and saddle points, the gradient of the energy is zero. Since the gradient is the negative of the forces, the forces are zero at such a stationary point. At a minimum, any alteration of the geometry increases the energy and hence, all normal vibrations are positive. At a saddle point, displacement along a particular normal vibration decreases the energy and hence the vibration has a negative frequency, but all other displacements result in an energy increase. All optimizations can locate a stationary point, although not always a global minima.
1.11 Geometry optimization using Gaussian03
The structure of the molecule can be optimized using one of the several quantum chemistry packages. The most popular among them is Gaussian03 [Frisch et al 2004]. The next section discusses the steps, as shown in the form of a flowchart in Fig. 1.5, to be followed for carrying out geometry optimization in Gaussian03.
1.11.1 Input Specification
For doing a calculation in Gaussian03, an input file is required. The input file contains the route section, information about the molecular charge and multiplicity, and definitions of the molecular structure given in Z-matrix. All this information is used to calculate the total number of electrons and the orbital occupancies. Shown in Fig. 1.6 below is a sample input file for geometry optimization of water molecule.
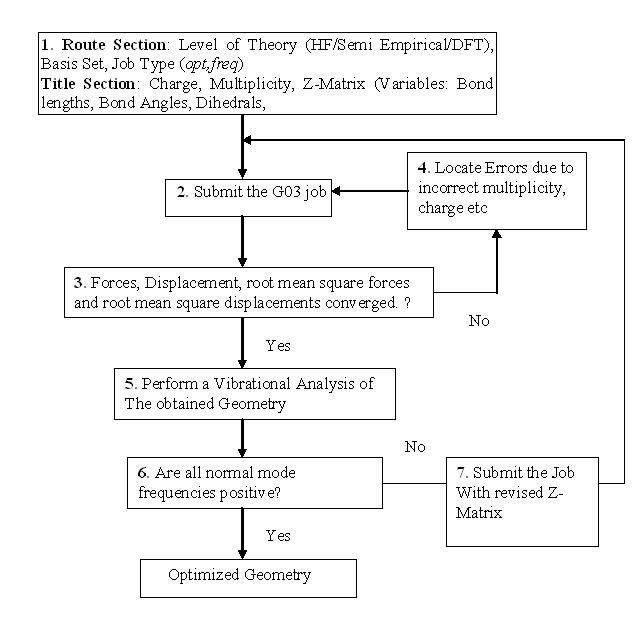
Figure 1.5 Procedure for geometry optimization
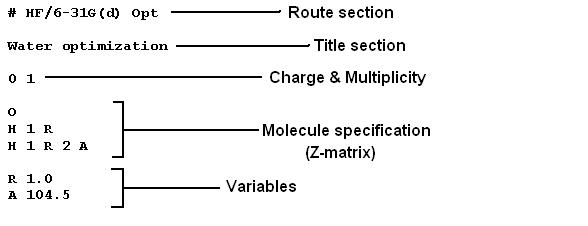
Sample input file for geometry optimization of water molecule