1.9 Working of the Gaussian program
The first task is to determine the type of calculation required. The GAUSSIAN program [Frisch et al., 2004] is usually controlled by specific keywords, which request given types of calculation. If the keywords are used, the program converts them to internal parameters, which then control the execution.
The various steps are summarized step by step:
- The first step is to read in a title for the job plus molecular charge, multiplicity (singlet, triplet, etc.),molecular geometry in the form of a Z-matrix. This consists of atomic numbers, bond lengths, bond angles, dihedral angles from which the Cartesian (x, y, z) coordinates are calculated. The information from the Z-matrix is then used to work out electronic configuration and the orbital occupancies.
- In the next step the nuclear repulsion energy which depends on the atomic numbers and the molecular geometry is calculated.
- The atomic orbitals or basis sets are then assigned to each nucleus. Ab-initio programs use an internally stored standard set of coefficients and exponents that define the orbitals (the basis set).
- Ab-initio programs next calculates the various one- and two-electron integrals required later in the calculation.
- Ab-initio programs then produce an initial guess of molecular orbitals used as a starting point for the SCF calculations. The usual form of initial guess for ab-initio programs is that obtained from an extended Hückel calculation [Wolfsberg and Helmholz, 1952] on the molecule in question.
- The solution to the SCF equations is iterated cycle by cycle until the electronic energy is at a minimum and the density matrix does not change. At this stage the calculation is said to be converged, or to have reached self consistency, and the program proceeds to the next step.
- For a geometry optimization the atomic forces are then determined analytically and used to estimate the minimum-energy geometry for the molecular species being calculated.
- The above process is repeated for each new geometry until the atomic forces are close to zero and the total energy does not change significantly from cycle to cycle.
- The next stage of the calculation depends on the type of job to be performed. For a single point, the program may either move directly to the population analysis, which calculates the atomic charges, overlaps, dipole and higher moments. The correlation energy for DFT is then calculated using Eqn 1.36.
At this stage the optimization is complete and the program moves on to a population analysis of the optimized species. Fig. 1.4 shows the flow chart for the typical ab-initio job.
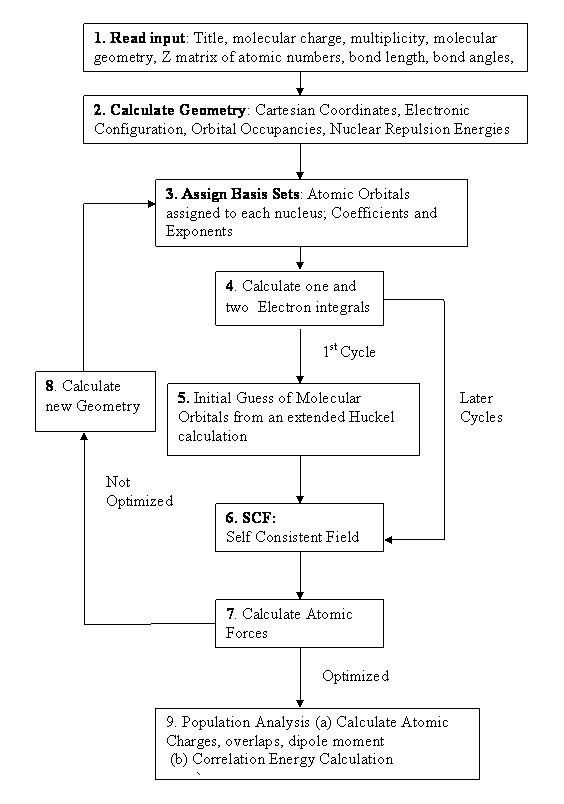
Figure 1.4 Typical flow chart for an ab-initio optimization