|
(1.27) |
|
(1.28) |
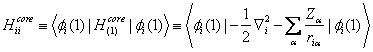
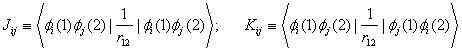
The one-electron core Hamiltonian ![]() omits the interactions of electron i with the other electrons. The sums over i and j are over the n/2 occupied spatial orbitals
omits the interactions of electron i with the other electrons. The sums over i and j are over the n/2 occupied spatial orbitals ![]() of the n-electron molecule. In the Coulomb integrals Jij and the exchange integrals Kij, the integration goes over the spatial coordinates of electrons 1 and 2. The Hartree-Fock method looks for those orbitals
of the n-electron molecule. In the Coulomb integrals Jij and the exchange integrals Kij, the integration goes over the spatial coordinates of electrons 1 and 2. The Hartree-Fock method looks for those orbitals ![]() that minimize the variational integral EHF. The molecular orbitals (MOs) are taken to be normalized and mutually orthogonal. The closed-subshell orthogonal Hartree-Fock MOs satisfy
that minimize the variational integral EHF. The molecular orbitals (MOs) are taken to be normalized and mutually orthogonal. The closed-subshell orthogonal Hartree-Fock MOs satisfy
|
(1.29) |
where εi is the orbital energy and where (Hartree-) Fock operator ![]() is
is
|
(1.30) |
|
(1.31) |
where the Coulomb operator![]() and the exchange operator
and the exchange operator![]() are defined by
are defined by
|
(1.32) |
|
(1.33) |
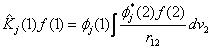
where f is an arbitrary function and the integrals are definite integrals over all space. In our considerations f implies an orbital.
The first term on the right of Eq. (1.31) is the kinetic energy operator for one electron; the second term is the potential-energy operators for the attractions between one electron and the nuclei. The Coulomb operator ![]() is the potential energy of interaction between electron 1 and a smeared-out electron j with electronic density
is the potential energy of interaction between electron 1 and a smeared-out electron j with electronic density ![]() ; the factor 2 occurs because there are two electrons in each spatial orbit. The exchange operator has no simple physical interpretation but arises from the requirement that the wave function be antisymmetric with respect to electron exchange. The Hartree-Fock Hamiltonian operator
; the factor 2 occurs because there are two electrons in each spatial orbit. The exchange operator has no simple physical interpretation but arises from the requirement that the wave function be antisymmetric with respect to electron exchange. The Hartree-Fock Hamiltonian operator ![]() is a one-electron operator (that is, it involves the coordinates of only one electron) and is peculiar in that it depends on its own eigen functions which are not known initially. Hence the Hartree-Fock equations must be solved by an iterative procedure known as the self-consistent field (SCF) method in which the orbitals are improved from cycle to cycle until the electronic energy reaches a minimum value and the orbitals no longer change. This situation is described as “self-consistent.”
is a one-electron operator (that is, it involves the coordinates of only one electron) and is peculiar in that it depends on its own eigen functions which are not known initially. Hence the Hartree-Fock equations must be solved by an iterative procedure known as the self-consistent field (SCF) method in which the orbitals are improved from cycle to cycle until the electronic energy reaches a minimum value and the orbitals no longer change. This situation is described as “self-consistent.”
In real systems the movements of the electrons are not independent of each other, as assumed in Hartree-Fock method, but are correlated to a certain extent so as to minimize repulsions as much as possible. This electron correlation means, in effect, that if electron A is at one end of the molecule, electron B prefers to be at the other end.