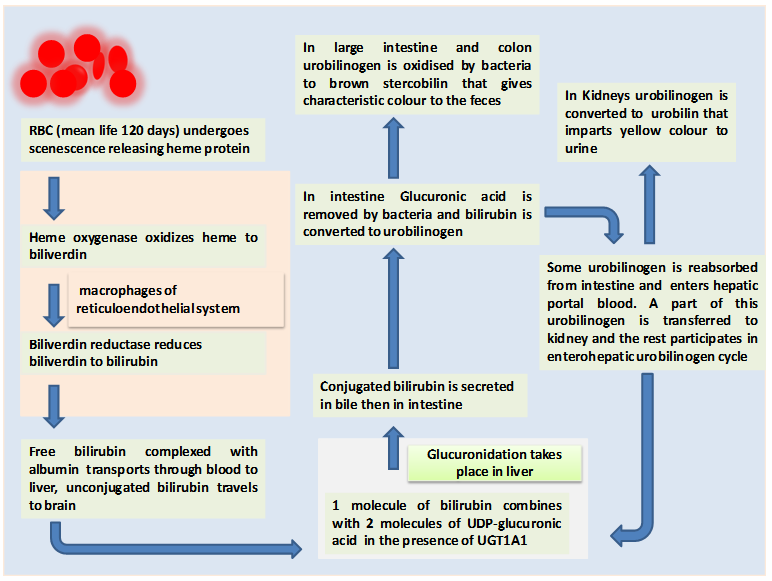
Figure 27.1 Metabolism of hemoglobin and glucuronidation reaction
Crigler-Najjar syndrome is a rare autosomal recessive disorder with an estimated case of 0.6–1.0 per million live births and is of two types. In type I patients the enzyme UGT1A1 is inactive whereas in type II it is severely reduced. In type I serum bilirubin level is usually above 345 µmol/L (310–755) which is very high as compared to normal serum bilirubin level of 2–14 μmol/L. Type I is difficult to treat as compared to type II. In type I the unconjugated bilirubin cannot be excreted into the bile and remains in the blood causing an elevated level of unconjugated bilirubin in plasma that leads to jaundice and may lead to kernicterus (bilirubin encephalopathy).
Long back when phototherapy was unavailable many children afflicted with Crigler-najjar syndrome died of kernicterus (bilirubin encephalopathy) some who survived till early adulthood developed neurological damage. The therapy includes
-
• 12h/d phototherapy (phototherapy becomes ineffective at later stage)
• Exchange transfusions of blood in the immediate neonatal period
• Use of heme oxygenase inhibitors
• Administration of oral calcium salts (help in complexes formation with bilirubin in the gastrointestinal tract)
• liver transplantation (Surgical intervention before the onset of brain damage and at later age when phototherapy becomes ineffective)