| |
1.4 Classical MC/MD Simulations (Module III) |
Using ab initio and semiempirical methods, one can determine intermolecular forces. This procedure is quite elaborate and for molecules containing more than 8 to 10 atoms, it is still customary to use empirical intermolecular potentials. These potentials are chosen in such a manner that the observed macroscopic properties of fluids consisting of these molecules are reproduced satisfactorily. |
For liquids of chemical and biological interest, it is still expedient to use classical equations of motion to calculate the molecular trajectories of all the particles in the chosen system. Since it is not possible to consider more than a few hundred to a few thousand molecules in a system, periodic boundary conditions are used to make the system resemble a bulk fluid. From the ensemble of particle positions and velocities that are generated, different thermodynamic functions such as the internal energy, heat capacity and pressure can be calculated. Of greater interest are the spatial and time correlation functions (TCFs). The spatial (radial) interparticle distribution functions (RDFs) can be compared with the experimental X-ray and neutron diffraction data. While a time correlation function (TCF) cannot be directly compared with experimental data, the transport coefficients (such as the diffusion coefficient, conductivity and viscosity) calculated using these TCFs can be used to validate the potential models used in performing the simulations. The development of a code for performing molecular dynamics for a Lennard-Jones fluid is considered in this module. The dynamical equations have to be solved using suitable algorithms. An alternative method to obtain the equilibrium properties of fluids is the Monte Carlo method. In this method, time does not appear explicitly explicitly during each step. Molecules are moved around their original locations by using a set of random numbers. The acceptance or rejection of such a move is decided by comparing the ratio of Boltzmann factors for the final and initial energies 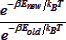 with another random number. If the ratio is greater than the random number, the new set of molecular coordinates is accepted, even if the new energy is higher than the old energy. If the new energy is lower than the old energy, the new configuration is automatically accepted. Steps in the development of the MC code will also be described in this module. with another random number. If the ratio is greater than the random number, the new set of molecular coordinates is accepted, even if the new energy is higher than the old energy. If the new energy is lower than the old energy, the new configuration is automatically accepted. Steps in the development of the MC code will also be described in this module. |
|
|
|