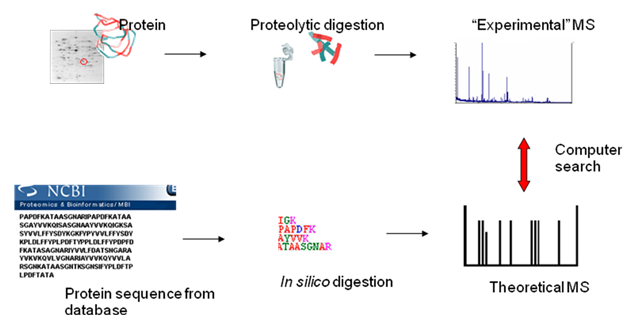
Figure 1: Various steps of Peptide mass fingerprinting
Although, the method looks very simple, there are certain limitations
- Peptide mass fingerprinting algorithms assumes the peptides come from a single protein. If there is any impurity the search result will not work.
- If protein has post translational modification, identification by Peptide mass fingerprinting does not work. As the methods assumes that the peptide mass is coming from amino acids only. Moreover, when genome data is virtually converted to protein, post-translational modification is difficult to precisely predict to match with experimental data.
- Although, specific proteases are available for protein cleave but sometime they cleave at wrong site (Mis-cleavage) which results in false prediction.
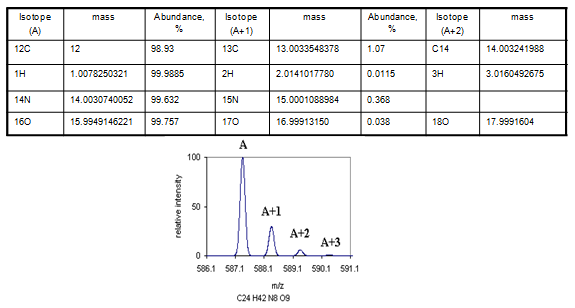
Each data peak in mass spectra may have more than one peak as atoms have several isotopes (Table 1). These peaks called isotopic peaks. For example a fragment of protein with mass M, when analyzed by mass spectrometry will give a major peak corresponding to mass M (Monoisotopic peak) but a small fraction will show M+1 mass because a small fraction of protein molecule may have 13C isotope. While searching database for protein identification (Fig. 2), one must mention the peak corresponds to monoisotopic or average.
Table 1: Isotopes of few common atoms